Inestabilidad genómica en mieloma múltiple
Genomic instability in multiple myeloma
Estela Pedrazzini1,2, Flavia Stella1,3,4, Irma Slavutsky1
1. Laboratorio de Genética de Neoplasias Linfoides, Instituto de Medicina Experimental, CONICET-Academia Nacional de Medicina, Buenos Aires, Argentina.
2. Departamento Ciencias Básicas y Experimentales, Universidad del Noroeste de la Provincia de Buenos Aires (UNNOBA), Buenos Aires, Argentina.
3. Área de Genética, Servicio de Anatomía Patológica, Hospital Posadas, Buenos Aires, Argentina.
4. Facultad de Ciencias Exactas, Químicas y Naturales, Universidad de Morón.
islavutsky@hematologia.anm.edu.ar; islavutsky@hotmail.com
Palabras clave: mieloma múltiple,
inestabilidad genómica,
firmas mutacionales,
epigenética
Keywords: multiple myeloma,
genomic instability,
mutational signatures,
epigenetics
Resumen
La inestabilidad genómica (IG) es una característica observada en casi todas las neoplasias y se define como una tendencia aumentada del genoma a adquirir diferentes tipos de cambios. El mieloma múltiple (MM) se caracteriza por una heterogeneidad genética importante evidenciada por numerosas alteraciones cromosómicas numéricas y estructurales recurrentes, primarias y secundarias, presentes al diagnóstico o adquiridas durante la evolución de la enfermedad, que sustentan una importante IG en la patología. Existen diferentes formas de IG que abarcan: inestabilidad cromosómica (IC), de microsatélites (IMS) y nucleotídica (IN). La IC constituye la forma más común en cáncer humano y se caracteriza por la existencia de una tasa acelerada de alteraciones cromosómicas durante las sucesivas divisiones celulares. Existen diferentes mecanismos relacionados al desarrollo de IC, entre ellos: rupturas del ADN a nivel de 1q12 asociada a la presencia de translocaciones “jumping”, cromotripsis, cromoplexia e inserciones con cambio de templado. A estas alteraciones corresponde agregar el acortamiento telomérico y el asincronismo de replicación. La IMS ha sido poco estudiada en MM. En cuanto a la IN, se caracteriza por sustituciones de base y pequeñas inserciones/deleciones (indels) que involucran tanto el genoma codificante como el no-codificante, generando firmas mutacionales distintivas que pueden afectar diferentes caminos de señalización. Entre ellas las más relevantes son: desaminación de metilcitosinas, APOBEC, actividad aberrante de AID y kataegis. Su presencia se asocia a progresión de la enfermedad y resistencia al tratamiento. Finalmente, cabe destacar la importancia de las alteraciones epigenéticas: metilación del ADN, modificaciones de histonas y expresión de ARNs no-codificantes, que presentan también un rol importante en el desarrollo neoplásico. Sin duda, el estudio de estos mecanismos de IG en el MM permitirá ahondar en el conocimiento de las características biológicas de la patología, pudiendo constituir un aporte en la generación de nuevas estrategias terapéuticas.
Abstract
Genomic instability (GI) is a characteristic present in most types of cancer, defined as an increased genome trend to acquired different types of changes. Multiple myeloma (MM) is characterized by an important genetic heterogeneity manifested by numerous recurrent numerical and structural cytogenetic alterations, observed at diagnosis or acquired during disease evolution, supporting an important GI in this pathology. There are different forms of GI: chromosome instability (CIN), microsatellite instability (MSI) and nucleotide instability (NI). CIN is the most common form in human cancer. It is characterized by an accelerated rate of chromosome alterations during cellular divisions. There are different mechanisms related to CIN development, among them: DNA breakpoints at 1q12 associated to jumping translocations, chromothripsis, chromoplexy and template insertions. In addition, telomere shortening and replication asynchrony must be considered. On the contrary, there is few information about MSI in MM. In reference to NI, it is characterized by base substitutions and small insertions/deletion (indels) that involve both the coding and non-coding genome, originating different mutational signatures that can affect distinct signaling pathways. The most relevant are: cytosine deaminases, APOBEC, AID aberrant activity and kataegis. They are associated to disease progression and resistance to treatment. Finally, it is important to point out the epigenetic alterations: DNA methylation, histone modifications and non-coding RNA expression, with an important role in neoplastic development. The study of GI mechanisms in MM will permit to deepen into the knowledge of the biologic characteristics of this pathology, making a contribution to the generation of novel therapies for these patients.
Inestabilidad genómica
La inestabilidad genómica (IG) es una característica presente en casi todos los tipos de cáncer, definiéndose como una tendencia aumentada del genoma a adquirir cambios(1,2). diversas modificaciones genéticas capaces de generar alteraciones de naturaleza temporaria o permanente en el genoma, cuya frecuencia, causas subyacentes y relevancia en la enfermedad varían significativamente entre las diferentes neoplasias(2-4). Existen distintas formas de IG que abarcan: inestabilidad cromosómica (IC), de microsatélites (IMS) y nucleotídica (IN), así como modificaciones epigenéticas que incluyen: metilación de ADN, modificaciones de histonas y expresión de ARNs no-codificantes (Tabla 1).
Tabla 1. Mecanismos de inestabilidad genómica
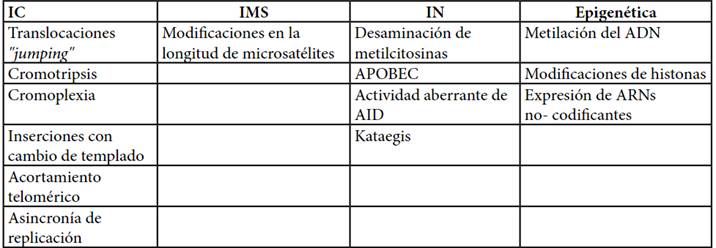
IC: inestabilidad cromosómica; IMS: inestabilidad de microsatélites; IN: inestabilidad nucleotídica; APOBEC (apolipoprotein B mRNA editing, catalytic polypeptide-like); AID: (activation-induced cytidine deaminases).
La IC constituye la forma más común en cáncer humano y se caracteriza por la existencia de una tasa acelerada de alteraciones cromosómicas durante las sucesivas divisiones celulares. Las mismas pueden resultar en ganancias o pérdidas de cromosomas enteros que varían dentro y entre las células tumorales (IC numérica), o en la generación de aberraciones estructurales no balanceadas, definidas como variaciones en el número de copias de regiones subcromosómicas (IC estructural)(5,6). Si bien se originan a través de diferentes mecanismos celulares, los mismos se encuentran relacionados entre sí de manera tal que la presencia de IC numérica puede inducir o acelerar IC estructural y viceversa(7,8). El fenotipo IC se encuentra relacionado a defectos en diferentes procesos como: reparación del ADN, control del ciclo celular, duplicación de los centrosomas, unión de los cromosomas a los microtúbulos, replicación, estabilidad de la telomerasa y modificaciones epigenéticas, entre otros(9). Asimismo, la IC contribuye a la transformación maligna, ya sea generando amplificación de oncogenes o pérdida de genes supresores de tumor, favoreciendo la progresión tumoral, recaída y resistencia al tratamiento(10,11).
IC en mieloma múltiple (MM)
El MM es una neoplasia de células B maduras post-centro germinal. Se caracteriza por una heterogeneidad genética importante evidenciada por numerosas alteraciones cromosómicas numéricas y estructurales recurrentes primarias y secundarias, presentes al diagnóstico o adquiridas durante la evolución de la enfermedad, que sustentan una importante IG(12-14). La acumulación de estas anomalías durante la progresión tumoral permite la expansión de clones resistentes, contribuyendo al desarrollo de enfermedad de alto riesgo(15,16).
Uno de los mecanismos relacionados con el desarrollo de IC en MM son las rupturas de ADN a nivel de 1q12, que se asocian a la presencia de translocaciones “jumping”, capaces de inducir un alto número de rearreglos transientes y clonales, particularmente ganancias de 1q21(17-20). Esta región corresponde a la ubicación de la heterocromatina pericentromérica del brazo largo del cromosoma 1 que evidencia una habilidad importante para duplicarse a sí misma y a regiones adyacentes generando alteraciones desbalanceadas con diferentes cromosomas receptores. Una posible causa de estas duplicaciones directas o inversas y de translocaciones “jumping” es la descondensación de la heterocromatina pericentromérica (secuencias SatII/III) ubicada en 1q12, que permitiría, mediante ciclos de ruptura y fusión (BFB; breakage-fusion-bridges), la recombinación y formación de trirradiales, isocromosomas, translocaciones desbalanceadas y micronúcleos conteniendo 1q. Si bien no se conoce la causa de esta alteración, se especula que podría estar relacionada a una hipometilación región-específica de 1q12(19,20), que contiene un sitio frágil y se asocia a expansiones de repeticiones de ADN satélite en diferentes neoplasias(21). En las células normales esta región permanece altamente condensada, pero en las células tumorales se descondensa, siendo propensa a rupturas y translocaciones “jumping”. Más recientemente se han sugerido otras modificaciones de la región 1q12, entre ellas una reprogramación epigenética del dominio de ADN satélite, que coincide con una demetilación global inducida por la inhibición de DNMT (DNA Methyltransferase)(22). Otra causa posible de esta inestabilidad se relaciona con la sobreexpresión de la enzima modificadora de la cromatina KDM4A (Lysine Demethylase 4a), una histona demetilasa que se une al locus BCL9 (BCL9 Transcription Coactivator) (1q21.2), causando replicación y aumento del número de copias de 1q12 y 1q21, alterando la expresión de microRNAs y la presión selectiva(23-25). Otros estudios han relacionado esta inestabilidad con alteraciones en el procesamiento del ARN a través de la amplificación y sobreexpresión del gen ILF2 (Interleukin Enhancer Binding Factor 2) que promueve tolerancia a la IG y la estabilización de transcriptos involucrados en recombinación homóloga, determinando un aumento de puentes nucleoplásmicos y la formación de micronúcleos(26,27), así como el aumento de expresión del gen ADAR1 (Adenosine Deaminase RNA Specific) (1q21.3) que actúa promoviendo la progresión del MM y sobrevida corta (SV)(28).
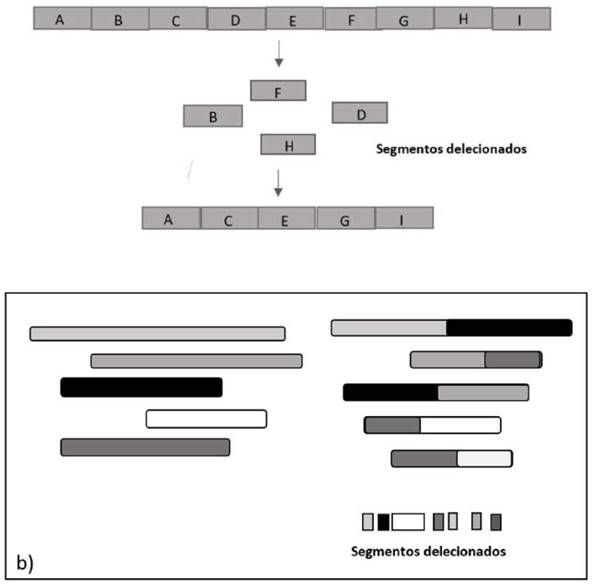
Otros mecanismos de IC relativamente nuevos detectados en pacientes con MM son cromotripsis, cromoplexia y ciclos de inserciones con cambio de templado(2). Cromotripsis (del griego Chomo: cromosoma y Thripsis: romperse en pedazos) se define como el fenómeno en el cual se producen decenas, e incluso cientos, de reordenamientos cromosómicos que ocurren en un solo evento catastrófico(29) (Figura 1a). Este proceso ha sido relacionado con la formación de cromosomas dobles diminutos, que representan amplificación oncogénica y se manifiestan como cientos de copias en una misma célula, visualizándose como múltiples microcromosomas sin centrómeros. También puede ocurrir como resultado de una segregación errónea de cromosomas dicéntricos originados a partir de fusiones teloméricas(30). El fenómeno de cromotripsis ocurre como un evento clonal único, presente en el 2-3% de los tumores primarios, y constituye un modelo alternativo en el desarrollo tumoral frente a la adquisición progresiva de mutaciones. Empleando la técnica de array de SPN (single nucleotide polimorphism) se observó la presencia de cromotripsis en el 1,3% de los pacientes con MM, los que representarían una entidad biológica diferente de alto riesgo(31), probablemente relacionada con la desregulación de un gran número de genes. Estudios más recientes mediante NGS (next generation sequencing)(32) permitieron observar la presencia de cromotripsis en el 36% de los genomas de MM analizados, siendo un evento temprano en su patogénesis. No obstante, en algunos casos se encontraron evidencias de cromotripsis tardía, asociada a progresión de la enfermedad. Por su parte, cromoplexia (del griego Chomo: cromosoma y pleko: tejer) es también un evento genético catastrófico que genera rearreglos desordenados de múltiples cromosomas (Figura 1b). Se origina a partir de la ocurrencia simultánea de rupturas de doble cadena en numerosos cromosomas, que se reordenan incorrectamente, produciendo una cadena de rearreglos estructurales(2)Implica menos reordenamientos y segmentos delecionados que la cromotripsis. S(32). En cuanto a los ciclos de inserciones con cambio de templado, es un mecanismo de reparación de la replicación del ADN en el que la ADN-polimerasa cambia repetidamente de templado obstaculizando la progresión de la horquilla de replicación, generando múltiples repeticiones concatenadas que originan alteraciones en el número de copias en numerosos cromosomas(2)(32)
Figura 1. Inestabilidad cromosómica: a) cromotripsis; b) cromoplexia.
Disfunción telomérica
Los telómeros son regiones de ADN no codificante ubicadas en los extremos de los cromosomas eucarióticos, constituidos por secuencias de ADN altamente conservadas, repetidas en tándem (TTAGGG) y asociadas a proteínas específicas. Cumplen un rol fundamental en la protección del ADN de la acción de enzimas degradativas y de las fusiones terminales, preservando la estabilidad e integridad cromosómica. Por otra parte, determinan interacciones importantes entre los cromosomas y la matriz nuclear, influyendo en la localización de los mismos en el núcleo, el apareamiento de los cromosomas homólogos y su movimiento durante la división celular. Asimismo, ejercen efectos sobre la transcripción de genes situados en regiones subteloméricas e interactúan con los mecanismos regulatorios del ciclo celular(33).
Los telómeros disfuncionales, críticamente acortados, determinan la activación de los mecanismos de respuesta del daño al ADN, siendo considerados una de las causas de IG(34). Dicho acortamiento telomérico aumenta la probabilidad de fusiones entre extremos cromosómicos, generando ciclos de BFB que resultan en rearreglos cromosómicas usualmente encontrados en diversos tipos de cáncer(35,36), asociado en algunos casos a la presencia de mutaciones en genes que codifican para proteínas que regulan la longitud telomérica (LT)(37-39).
En este contexto, el mantenimiento de la LT es de suma importancia en la tumorigénesis y la inmortalización celular, y se encuentra fuertemente implicado en el origen de IC. El mismo depende de la interacción entre la enzima telomerasa y la maquinaria de replicación del ADN. La telomerasa es un complejo ribonucleoproteico constituido por dos subunidades: TERT (Telomerase Reverse Transcriptase) que presenta actividad catalítica de transcriptasa reversa, y TERC (Telomerase RNA Component) que provee el molde para la adición de nuevas repeticiones teloméricas al extremo 3´ simple cadena(40,41). En humanos, la telomerasa se encuentra ausente en la mayoría de las células somáticas normales, presenta niveles bajos en poblaciones celulares con alto potencial proliferativo como linfocitos activados y células de las criptas intestinales, entre otras, y muestra altos niveles de expresión en células germinales, madre y tumorales, así como en líneas celulares inmortalizadas(42). La estabilización de la LT por activación de la telomerasa se observa en el 85% de todas las formas de cáncer humano, sugiriendo un rol para esta enzima durante la progresión tumoral. Recientemente, el hallazgo de mutaciones en la región promotora del gen TERT en tejidos tumorales con bajo índice de replicación, provee un nuevo mecanismo de reactivación de la telomerasa en cáncer. Se cree que estas mutaciones generan un sitio de unión para el factor de transcripción ETS (E-twenty-six), causando un aumento en la expresión de TERT y afectando a la LT(43).
Diferentes trabajos han evaluado la LT y la actividad de telomerasa en pacientes con MM(44-49). Distintos autores(46,47) muestran la presencia de acortamiento telomérico en MM y su asociación con cariotipos anormales, asociaciones teloméricas y aumento de la actividad de telomerasa, que definiría a un subgrupo de pacientes con mal pronóstico, sustentando la importancia de este mecanismo en el desarrollo y progresión de la enfermedad(47,49). En concordancia con estos datos, Hyatt et al(48) demostraron, a través de un modelo multivariado, que la LT y la edad serian factores pronóstico importantes a incluir en el ISS (International Staging System) para una mejor estadificación del MM, considerando a la LT como un determinante crítico de la SV en esta patología. Datos de nuestro grupo muestran también asociación entre acortamiento telomérico e integrantes de los complejos protector (shelterin) y no protector (no-shelterin) de los telómeros. Particularmente, Panero et al(50,51) encuentran asociación inversa entre la expresión de TRF2 (Telomeric Repeat Binding Factor 2), TANK1 (Tankyrase) y DKC1 (Dyskerin 1) con la LT, sustentando la participación de otros mecanismos, además de la telomerasa, en el mantenimiento del tamaño e integridad telomérica en MM, y sus implicancias en el desarrollo de IG.
Asincronismo de replicación (AR)
Como sabemos, la replicación del genoma es un proceso esencial que garantiza la copia precisa de la información genética antes de la división celular. Cada ciclo de replicación representa una oportunidad de error que conduce a la adquisición de mutaciones y de alteraciones del número de copias(52). La sincronización del programa de replicación del ADN es un proceso altamente organizado con algunos locus de replicación temprana y otros de replicación tardía durante la fase S del ciclo celular(53). Asimismo, existe una asociación estrecha entre el intervalo específico de tiempo durante la fase S en el cual una secuencia particular de ADN se replica en un determinado tejido y su actividad transcripcional, considerándose de replicación temprana los loci que se están expresando en ese tejido y de replicación tardía los que no se expresan(54). Diferentes estudios muestran que las neoplasias están acompañadas de una disrupción del orden temporal de la replicación alélica, observándose que algunos genes implicados en el desarrollo maligno replican sincrónicamente en células diploides normales, en tanto que evidencian un marcado grado de asincronismo en células neoplásicas(55,56). Dicha AR, entendida como la pérdida temporal del control de la replicación, puede generar cambios genéticos como aneuploidías y epigenéticos, como inactivación alélica, equivalente a la pérdida de heterocigocidad, que llevarían al silenciamiento de genes supresores de tumor o a la activación oncogénica(55-57). La AR puede ser detectada mediante la técnica de FISH (Fluorescence in situ hybridization), constituyendo una herramienta sensible para evaluar el momento de replicación de segmentos específicos del genoma, haciendo factible estimar la sincronización de la replicación de un alelo respecto de su contraparte en una misma célula. Permite distinguir una secuencia de ADN no replicada en un núcleo interfásico como una señal fluorescente única (S) mientras que la secuencia replicada se visualiza como una señal doble (D). De esta manera, en una población de células en división, una frecuencia alta de núcleos con dos señales de hibridación similares indicaría que el par de alelos replica sincrónicamente, en tanto que aquellos núcleos que contienen dos señales de hibridación diferentes (SD) indican la presencia de AR (Figura 2).
Existe poca información en la literatura respecto de la presencia de AR en pacientes con desórdenes de células plasmáticas. Amiel et al(58) evaluaron AR en los loci TP53 (17p13), RB1 (13q14) y 21q22 en pacientes con MM y MGUS (monoclonal gammopathy of undetermined significance) encontrando un aumento significativo del porcentaje de células asincrónicas en MM respecto de controles, con valores intermedios para el MGUS, indicando un rol para la AR en la progresión de MGUS a MM. Un trabajo reciente de nuestro laboratorio(59), analizó la AR en los loci TP53 y RB1 en pacientes con MM detectando un aumento significativo del porcentaje de células asincrónicas en los casos con deleción de estos genes y de pacientes con cariotipo anormal respecto de aquéllos con cariotipo y FISH normal y de controles, sustentando una asociación entre las modificaciones en la AR y la adquisición de alteraciones genéticas e IG en esta patología.
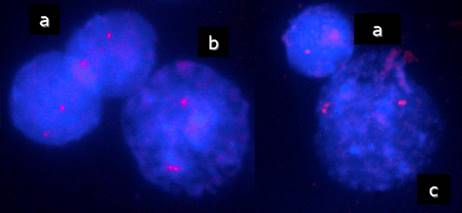
Figura 2. Núcleos interfásicos hibridados con la sonda RB1 13q14 (Live-Lexel, Buenos Aires, Argentina) mostrando asincronismo de replicación: a) replicación sincrónica temprana: dos señales simples (SS); b) replicación asincrónica: una señal simple y una doble (SD); c) replicación sincrónica tardía: dos señales dobles (DD).
Inestabilidad de microsatélites (IMS)
La IMS es otra forma de IG, presente en las diferentes neoplasias. Los microsatélites son secuencias cortas de ADN, de uno a seis pares de bases, repetidas en tándem a lo largo del genoma eucariota. Se caracteriza por una disminución o un aumento en la longitud de los microsatélites en el ADN tumoral en comparación con el ADN normal, y se asocia a una mayor frecuencia de mutaciones puntuales(60,61). Esta alteración puede ser corregida por las enzimas codificadas por los genes de reparación del ADN (MMR: mismatch repair), siendo considerada un desorden de los genes MMR, que impide que los errores de replicación cometidos por la ADN-polimerasa sean reparados, determinando una mayor tasa de mutación, particularmente en las regiones de microsatélites, y la aparición de un fenotipo mutador.
IMS ha sido relativamente poco estudiado en MM. Un primer reporte de Velangi et al(62) detectó su presencia en el 7,7% de los casos con MGUS/MM asintomático, en el 20,7% en aquéllos con MM sintomático/leucemia de células plasmáticas (LCP) y en el 12,5% de los pacientes en recaída, sugiriendo un incremento de la IG durante la progresión de la enfermedad. Por su parte, Timurağaoğlu et al(63) observaron IMS en el 54% de los pacientes con MM al momento del diagnóstico, aunque sin encontrar asociación con factores pronóstico de la patología. Un trabajo más reciente(64) empleando la técnica HRFMA (high resolution fluorescent microsatellite analysis), detectó una frecuencia mucho menor (10%) en pacientes con MM, siendo en todos los casos alteraciones de tipo A (con cambios de hasta 6 pb)(65). Si bien, el número de pacientes evaluados al presente es limitado, los resultados obtenidos muestran que el fenotipo mutador está presente en el MM, siendo necesario más estudios para confirmar estos hallazgos.
Eventos mutacionales asociados a IG
Además de los mecanismos previamente expuestos, en MM también resulta de importancia la IN, caracterizada por sustituciones de base y pequeñas inserciones/deleciones (indels) que involucran tanto el genoma codificante como el no-codificante(66). En algunos casos estas mutaciones generan firmas mutacionales distintivas que pueden afectar diferentes caminos de señalización, cuyo desarrollo lleva a progresión de la enfermedad y promueve la resistencia al tratamiento. La introducción de las técnicas de NGS junto con la aplicación de algoritmos computacionales modernos ha permitido ahondar en su conocimiento en el MM, habiéndose identificado al menos 17 firmas, algunas de las cuales resultan de particular importancia en relación al desarrollo de IG. Entre ellas cabe destacar:
1) Desaminación de metilcitosinas, caracterizada por estar enriquecida en transiciones C>T (sustitución de nucleótidos de la misma naturaleza química) en el contexto de dinucleótidos CpG, que ocurren como resultado de la desaminación de citosinas metiladas a timinas. Es frecuente en numerosos tipos de neoplasias y se la observa con alta frecuencia en los pacientes con MM(67-69).
2) APOBEC (apolipoprotein B mRNA editing, catalytic polypeptide-like) es una familia de enzimas de edición del ADN que actúan fundamentalmente sobre el ADN simple cadena a través de la desaminación de citosina a uracilo(80), generando mutaciones que pueden ser oncogénicas. Esta firma molecular tiene un patrón característico que resulta en el enriquecimiento de sustituciones C>T y C>G en un contexto de trinucleótidos TpCpA, y ha sido descripta en numerosos tumores sólidos y neoplasias hematológicas, entre ellas la leucemia linfocítica crónica(67,81). En MM el 3,8% de los pacientes presenta esta característica, asociada a las translocaciones que involucran a los genes MAF (MAF BZIP Transcription Factor): t(14;16)(q32;q23) y t(14;20)(q32;q11), con sobreexpresión de las isoformas APOBEC3A y APOBEC3B, respectivamente, y a una carga mutacional alta(69). Esta firma mutacional es de adquisición tardía(72,73) y se asocia a pronóstico adverso con SV libre de progresión y global cortas(74). Un trabajo reciente(75) observa un incremento de la actividad de APOBEC durante la progresión de las diferentes fases de los desórdenes de células plasmáticas, desde el MGUS/MM indolente al MM sintomático y la LCP primaria, con valor pronóstico adverso independiente. Asimismo, estos autores detectaron un 23% de casos con aumento de actividad de APOBEC, particularmente APOBEC3B, que no presentaban translocaciones que involucraran a los genes MAF, indicando la participación de otros factores en la modulación de este proceso aberrante. Estos hallazgos sugieren que el análisis de la actividad de APOBEC al momento del diagnóstico podría ayudar a identificar pacientes de riesgo alto que se beneficiarían con tratamientos más específicos.
3) Actividad aberrante de AID (activation-induced cytidine deaminases). AID es miembro de la familia de ADN deaminasas APOBEC, es una enzima esencial en la activación de los linfocitos B a nivel del centro germinal, responsable de los procesos de SHM (somatic hypermutation) y CSR (class switch recombination) de los genes de Igs, generando cambios de citosina a uracilo en la región variable de la cadena pesada de las Igs y cambio de isotipo de IgM a IgG o IgE(76). AID también puede producir mutaciones en otros genes, y su activación aberrante es capaz de generar activación oncogénica, rupturas del ADN de doble cadena, translocaciones e IG(77). Particularmente en MM, está involucrada en la translocación t(11;14)(q13;q32) que determina la yuxtaposición de los genes CCND1 (ciclina D1) e IGH (Immunoglobulin Heavy Locus)(69), teniendo un rol importante en la adquisición temprana de mutaciones líderes en esta patología(72,73,78). Bolli et al(72) detectaron un rol importante de AID en la progresión de MM indolente a sintomático, así como la presencia de una nueva firma mutacional que involucra la participación de la vía no canónica de AID observada en el 28% de los genomas analizados, más prevalente en las regiones no codificantes, y también de adquisición temprana. Estos datos resultan de importancia en la patogénesis del MM, así como también en la toma de decisiones terapéuticas.
4) Kataegis (del griego: καταιγίς, tempestad), definida como un patrón localizado de hipermutaciones focales en regiones adyacentes a rearreglos genómicos. Se caracteriza por la presencia de clústeres de transiciones C>T y/o C>G en el contexto de trinucleótidos TpCpN, que se producen sobre la misma cadena de ADN(67). Los focos pueden ser unos pocos a varios miles. Si bien no está totalmente clarificado, se considera que uno de los miembros de la familia APOBEC estaría involucrado en la generación de este mecanismo. En MM se observó asociación con las translocaciones que involucran principalmente al oncogén MYC, pero también se detectó en rearreglos de los cromosomas 1, 10, 11, 16 y 17(69). Se considera un evento temprano asociado a la actividad aberrante de las vías canónica y no canónica de AID(72,73).
Alteraciones epigenéticas
Además de las alteraciones genéticas características del MM, las modificaciones epigenéticas juegan también un rol importante en el desarrollo y progresión de la enfermedad. Dichas modificaciones constituyen cambios heredables en la expresión génica que se producen sin modificaciones en la secuencia de ADN(79). Los mecanismos más comunes de regulación epigenética son la metilación del ADN, las modificaciones post-traduccionales de las histonas y la expresión de ARNs no-codificantes. La metilación del ADN consiste en una modificación covalente postreplicativa que agrega un grupo metilo al anillo de citosina formando 5-metil citosina. La misma se produce sólo en las citosinas que preceden a guaninas, formando el dinucleótido CpG, que se agrupa en regiones pequeñas de 0,5-4 Kb, denominadas islas CpG que, a menudo, involucran a los promotores génicos, usualmente no metilados. La falta de metilación en las islas CpG permite la expresión del gen. La metilación aberrante del ADN es característica de las células tumorales humanas, particularmente la hipermetilación de los promotores de los genes supresores de tumor, que determina el silenciamiento génico, y la hipometilación del ADN asociada a IG. Diferentes estudios han mostrado que el MM se caracteriza por alteraciones en la metilación del ADN que son específicas en diferentes estadíos de la enfermedad(80-82). Concretamente, se observó una hipometilación global en MGUS y una progresiva hipermetilación en el MM sintomático y en los pacientes en recaída, sugiriendo que las modificaciones en el patrón de metilación contribuirían a la patogénesis de esta enfermedad(83-85). En este aspecto, resulta de interés el rol del gen MAFB, involucrado en la t(14;20). Estudios en ratones transgénicos con una neoplasia de células plasmáticas asociada a alta expresión de MAFB mostraron una reprogramación del perfil de metilación de las células madre hematopoyéticas/células progenitoras, que se mantenía en las células plasmáticas tumorales, demostrando un nuevo mecanismo molecular de la carcinogénesis(86). Por otra parte, Walker et al(81) reportaron una alta hipermetilación en los pacientes que presentaban la t(4;14) respecto de los otros grupos citogenéticos. Estos casos sobreexpresan MMSET (Multiple Myeloma SET Domain-Containing Protein), que tiene actividad de histona metiltransferasa e interactúa con co-represores tipo HDACs (histone deacetylases) regulando la transcripción génica(87). La sobreexpresión MMSET lleva a la metilación de H3K36 y H3K27 y regula el ensamblaje de 53BP1 (p53-binding protein 1) a los sitios de lesiones del ADN(88). De esta manera, la sobreexpresión debida a la t(4;14) determina modificaciones de histonas que promueven la sobrevida celular, la progresión del ciclo celular y causan una respuesta aberrante al daño al ADN, alterando la maquinaria de reparación y promoviendo la IG. Asimismo, Walker et al(81) observan un aumento de metilación a nivel de los promotores de diferentes genes en la transición de MM a LCP, sustentando la participación de este mecanismo en la progresión de la enfermedad.
Concluyendo, la IG contribuye a generar diferentes características del MM, como su complejidad genética, heterogeneidad clonal y subclonal, promoviendo resistencia al tratamiento. Muchos de estos estudios resultan factibles de realizar en nuestro medio siendo importante su desarrollo tendiente a una mejor caracterización biológica de la patología. Sin duda, el análisis de la participación de los diferentes mecanismos de IG en la evolución del MM permitirá ahondar en el conocimiento de esta entidad, así como constituir un aporte en la generación de nuevas estrategias terapéuticas.
Agradecimientos
El presente trabajo fue sustentado por la Universidad de Morón mediante Proyectos de Investigación Interinstitucionales y el Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
Bibliografía
1. Lee JK, Choi, Kwon, ParkMechanisms and consequences of cancer genome instability: lessons from genome sequencing studies. Annu Rev Pathol. 2016;11:283-312.
2. Alagpulinsa, SzalatRE, Poznansky, Shmookler ReisGenomic instability in multiple mieloma. Trends Cancer. 2020;10:858-73.
3. Mateuca R, Lombaert N, Aka PV, Decordier I, Kirsch-Volders M. Chromosomal changes: induction, detection methods and applicability in human biomonitoring. Biochimie. 2006;88:1515-31.
4. Negrini S, Gorgoulis VG, Halazonetis T D. Genomic instability – an evolving hallmark of cancer. Nature Rev Mol Cell Biol. 2010;11:220-8.
5. Roschke AV, Rozenblum E. Multi-layered cancer chromosomal instability phenotype. Front Oncol. 2014;3:302.
6. Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. The mitotic origin of chromosomal instability. Curr Biol. 2014;24:R148-9.
7. Crasta KK, Ganem NJ, Dagher RR y col. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53-8.
8. Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014;4:1281-9.
9. Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122:138-43.
10. Bakhoum SF, Landau DA. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb Perspect Med. 2017;7:a029611.
11. Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol. 2018;15:139-50.
12. Fonseca R, Bergsagel PL, Drach J y col. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210- 21.
13. Manier S, Salem, Park, Landau, Getz, Ghobrial. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017;14:100-13.
14. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93:1091-110.
15. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335-348.
16. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306-13.
17. Sawyer JR, Tricot G, Lukacs JL et al. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chrom Cancer. 2005;42:95-106.
18. Sawyer JR, Tian E, Heuck CJ y col. Jumping translocations of 1q12 in multiple myeloma: a novel mechanism for deletion of 17p in cytogenetically defined high-risk disease. Blood. 2014;123:2504-12.
19. Sawyer JR, Tian, Heuck. Evidence of an epigenetic origin for high-risk 1q21 copy number aberrations in multiple myeloma. Blood. 2015;125:3756-9.
20. Sawyer JR, Tian E, Walker BA y col. An acquired high-risk chromosome instability phenotype in multiple myeloma: Jumping 1q Syndrome. Blood Cancer J. 2019;9:62.
21. Bersani F, Lee E, Kharchenko PV y col. Pericentromeric satellite repeat expansions through RNA derived DNA intermediates in cancer. Proc Natl Acad Sci USA. 2015;112:15148-53.
22. Brückmann NH, Pedersen CB, Ditzel HJ, Gjerstorff MF. Epigenetic reprogramming of pericentromeric satellite DNA in premalignant and malignant Lesions. Mol Cancer Res. 2018;16:417-27.
23. Black JC, Manning, Van Rechem y col. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell. 2013;154:541-55.
24. Black JC, Atabakhsh, Kim. Hypoxia drives transient site-specific copy gain and drug resistant gene expression. Genes Dev. 2015;29:1018-31.
25. Black JC, Zhang H, Kim J, Getz G, Whetstine JR. Regulation of transient site-specific copy gain by microRNA. J Biol Chem. 2016;291:4862-71.
26. Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NS. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009; 113: 2290-7.
27. Marchesini M, Ogoti, Fiorini. ILF2 is a regulator of RNA splicing and DNA damage response in 1q21-amplified multiple myeloma. Cancer Cell. 2017;32:88-100.
28. Lazzari E, Mondala PK, Santos ND y col. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 2017;8:1922.
29. Stephens PJ, Greenman CD, Fu B y col. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27-40.
30. Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641-54.
31. Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood. 2011;118:675-8.
32. Maura F, Bolli N, Angelopoulos N y col. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019;10:3835.
33. Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921-9.
34. Gisselsson D, Jonson T, Petersén A y col. Telomere dysfunction triggers extensive DNA fragmentation and evolution to complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci USA. 2001;98:12683-8.
35. Gisselsson D, Jonson T, Yu C y col. Centrosomal abnormalities, multipolar mitoses, and chromosomal instability in head and neck tumours with dysfunctional telomeres. Br J Cancer. 2002;87: 202-7.
36. Höglund M, Gisselsson D, Hansen GB, Sähl T, Mitelman F. Ovarian carcinoma develops through multiple modes of chromosomal evolution. Cancer Res. 2003;63:3378-85.
37. Shay JW, Zou Y, Hiyama E, Wright WE. Telomerase and cancer. Hum Mol Genet. 2001;10:677-85.
38. Karlseder J, Smogorzewska A, De Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446-9.
39. Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068-71.
40. Bodnar AG, Ouellette M, Frolkis M y col. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349-52.
41. Cairney CJ, Keith WN. Telomerase redefined: Integrated regulation of hTR and hTERT for telomere maintenance and telomerase activity. Biochimie. 2008;90:13-23.
42. Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111: 4446-55.
43. Heidenreich B, Kumar R. TERT promoter mutations in telomere biology. Mutat Res. 2017;771:15-31.
44. Xu D, Zheng C, Bergenbrant S y col. Telomerase activity in plasma cell dyscrasias. Br J Cancer. 2001;84:621-5.
45. Shiratsuchi M, Muta K, Abe Y y col. Clinical significance of telomerase activity in multiple myeloma. Cancer. 2002;94:2232-8.
46. Cottliar A, Pedrazzini E, Corrado C, Engelberger MI, Narbaitz M, Slavutsky I. Telomere shortening in patients with plasma cell disorders. Eur J Haematol. 2003;71:334-40.
47. Wu KD, Orme LM, Shaughnessy J Jr, Jacobson J, Barlogie B, Moore MA. Telomerase and telomere length in multiple myeloma: correlations with disease heterogeneity, cytogenetic status, and overall survival. Blood. 2003;101:4982-9.
48. Hyatt S, Jones RE, Heppel NH y col. Telomere length is a critical determinant for survival in multiple myeloma. Br J Haematol. 2017;178:94-8.
49. Aref, Al Saeed, El Menshawy N, Abdalla, El Ashery MPrognostic relevance of telomere length and telomerase reverse transcriptase variant (rs2242652) on the multiple myeloma patients. J Clin Lab Anal. 2020;34:e23133.
50. Panero J, Arbelbide J, Fantl DB, García Rivello H, Kohan D, Slavutsky I. Altered mRNA expression of telomere-associated genes in monoclonal gammopathy of undetermined significance and multiple myeloma. Mol Med. 2010;16:471-8.
Differential Expression of Non-Shelterin Genes Associated with High Telomerase Levels and Telomere Shortening in Plasma Cell Disorders. PLoS One. 2015;10:e0137972.
52. De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat Biotechnol. 2011;29:1103-8.
53. Rhind N, Gilbert DM. DNA replication timing. Cold Spring Harb Perspect Med. 2013;3:1-26.
54. Goren A, Cedar H. Replicating by the clock. Nature Rev. 2003;4:25-32.
55. Dotan ZA, Dotan A, Litmanovich T y col. Modification in the inherent mode of allelic replication of lymphocytes of patients suffering from renal cell carcinoma: a novel genetic alteration associated with malignancy. Gene Chrom Cancer. 2000;27:270-77.
56. Korenstein-Ilan A, Amiel A, Lalezari S, Lishner M, Avivi L. Allele-specific replication associated with aneuploidy in blood cells of patients with hematologic malignancies. Cancer Genet Cytogenet. 2002;139:97-103.
57. Du Q, Bert SA, Armstrong NJ y col. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nature Comm. 2019;10:416.
58. Amiel A, Kirgner I, Gaber E, Manor Y, Fejgin M, Lishner M. Replication pattern in cancer: asynchronous replication in multiple mieloma and in monoclonal gammopathy. Cancer Genet Cytogenet. 1999;108:32-7.
59. Stella F, Pedrazzini E, Slavutsky I. Análisis citogenético y asincronía de replicación en pacientes con deleción 6q en mieloma múltiple. J Basic & Appl Genet. 2019;XXX Nº 1 (Suppl):98.
60. Leach FS, Nicolaides NC, Papadopoulos N y col. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215-25.
61. Li YC, Korol AB, Fahima T y col. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11:2453-65.
62. Velangi MR, Matheson EC, Morgan GJ y col. DNA mismatch repair pathway defects in the pathogenesis and evolution of myeloma. Carcinogenesis. 2004;25:1795-1803.
63. Timurağaoğlu A, Demircin S, Dizlek S, Alanoğlu, Kiriş. Microsatellite instability is a common finding in multiple myeloma. Clin Lymphoma Myeloma. 2009;9:371-4.
64. Miyashita K, Fujii K, Suehiro Y y col. Heterochronous occurrence of microsatellite instability in multiple myeloma – an implication for a role of defective DNA mismatch repair in myelomagenesis. Leuk Lymphoma. 2018;59:2454-9.
65. Oda S, Maehara Y, Ikeda Y y col. Two modes of microsatellite instability in human cancer: differential connection of defective DNA mismatch repair to dinucleotide repeat instability. Nucleic Acids Res. 2005;33:1628-36.
66. Janz S, Zhan F, Sun F y col. Germline risk contribution to genomic instability in multiple myeloma. Front Genet. 2019;10:424.
Hoang PH, Cornish AJ, Dobbins SE, Kaiser M, Houlston RS. Mutational processes contributing to the development of multiple myeloma. Blood Cancer J. 2019;9:60.
68. Bolli N, Avet-Loiseau, Wedge. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997.
69. Walker BA, Wardell, Murison. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997.
70. Bacolla A, Cooper DN, Vasquez KM. Mechanisms of base substitution mutagenesis in cancer genomes. Genes. 2014;5:108-46.
71. Rebhand, S, Huemer M, Gassner FJ y col. APOBEC3 signature mutations in chronic lymphocytic leukemia. Leukemia. 2014;28:1929-32.
72. Bolli N, Maura F, Minvielle S y col. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. 2018;9:3363.
73. Rustad EH, Yellapantula V, Leongamornlert D y col. Timing the initiation of multiple myeloma. Nat Commun. 2020;11:1917.
Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95:548-67.
75. Maura F, Petljak, Lionetti. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia. 2018; 32:1044-8.
76. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553-63.
77. Robbiani DF, Bunting S, Feldhahn N y col. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631-41.
78. Maura F, Rustad, Yellapantula. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia. 2019;34:1476- 80.
79. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683-92.
DNA methylation analysis of tumor suppressor genes in monoclonal gammopathy of undetermined significance. Ann Hematol. 2010;89:191-9.
81. Walker BA, Wardell CP, Chiecchio L y col. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117:553-62.
82. Heuck CJ, Mehta J, Bhagat T y col. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J Immunol. 2013;190:2966-75.
83. Moreaux J, Reme T, Leonard W y col. Development of gene expression-based score to predict sensitivity of multiple myeloma cells to DNA methylation inhibitors. Mol Cancer Ther. 2012;11:2685-92.
The epigenome in multiple myeloma: Impact on tumor cell plasticity and drug response. Front Oncol. 2018;8:566.
Epigenetics in multiple myeloma: From mechanisms to therapy. Semin Cancer Biol. 2018;51:101-15.
86. Vicente-Duenas C, Romero-Camarero I, Gonzalez-Herrero I y col. A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J. 2012;31:3704-17.
87. Martinez-Garcia E, Popovic R, Min DJ y col. The MMSET histone methyltransferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211-20.
88. Pei H, Zhang L, Luo K y col. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124-8.