Tumor de tejidos blandos como presentación atípica de linfoma de Burkitt esporádico. Reporte de caso
Soft tissue tumor as an atypical presentation of sporadic Burkitt’s lymphoma. A case report
Araúz-Valdés E1, Fernández-Castañeda L1, D’Orcy E2
1Servicio de Medicina Interna, Complejo Hospitalario Doctor Arnulfo Arias Madrid. Panamá, Panamá.
2Servicio de Hematología, Complejo Hospitalario Doctor Arnulfo Arias Madrid. Panamá, Panamá.
Dr Edwin Araúz = https://orcid.org/0000-0002-5668-4443
Dra Laura Fernández = https://orcid.org/0000-0002-8127-1640
Dra Elizabeth D´Orcy = https://orcid.org/0000-0003-2123-8686
Palabras claves: linfoma de Burkitt,
tejidos blandos,
tumor.
Keywords: Burkitt’s lymphoma,
soft tissue,
tumor.
Resumen
El linfoma de Burkitt es un linfoma de células B altamente agresivo. En adultos, la variante esporádica es poco frecuente, y su manifestación usualmente es en vísceras abdominales. La presentación clínica en tejidos blandos es extremadamente rara, con pocos casos reportados en la literatura. Son tumores agresivos por su alto índice de proliferación, siendo el síndrome de lisis tumoral espontánea una de las complicaciones metabólicas más frecuentes.
Presentamos el caso de un varón de 57 años sin antecedentes personales patológicos que debuta con masa de rápido crecimiento en región inguinal derecha que desarrolla síndrome de lisis tumoral espontáneo previo al tratamiento.
Abstract
Burkitt's lymphoma is a highly aggressive B-cell lymphoma. In adults, the sporadic variant is rare, and its manifestation is usually in abdominal viscera. Clinical presentation in soft tissue is extremely rare, with few cases reported in the literature. They are aggressive tumors due to their rapid proliferation, with spontaneous tumor lysis syndrome being one of the most frequent metabolic complications.
We present the case of a 57-year-old male without medical history with a rapidly growing mass in the right inguinal region who develops sporadic tumor lysis syndrome before treatment.
Introducción
El linfoma de Burkitt es un linfoma no Hodgkin (LNH) de células B caracterizado por rápido crecimiento y manifestaciones en múltiples sistemas. Representa menos del 5% de todos los linfomas en el adulto y se clasifica en tres entidades clínicas: endémica, esporádica y asociada a inmunodeficiencia. La variante esporádica representa menos del 1% de los LNH en la población adulta, y está asociado en un 20% al virus de Epstein Barr. El abdomen es el sitio anatómico más afectado, particularmente la región ileocecal. Su presentación en tejidos blandos es extremadamente rara. Debido a su alto índice de proliferación presenta alta mortalidad; sin embargo, al ser tratado de manera oportuna la tasa de supervivencia es favorable(1,2).
Caso clínico
Varón de 57 años que acude por aumento progresivo de volumen de miembro inferior derecho a nivel proximal, asociado a parestesia y claudicación intermitente de dos semanas de evolución. Posteriormente, desarrolla ictericia de tegumentos por lo que acude a atención médica.
Sin antecedentes personales patológicos y no patológicos.
Al examen físico presenta ictericia generalizada. Se palpa lesión pétrea, tipo masa, de aproximadamente 20 cm en región inguinal derecha, adherida a planos profundos y asociada a piel de naranja, edema escrotal y de miembro inferior ipsilateral.
A su ingreso sin alteración en el hemograma y función renal. Laboratorios con serología viral para VIH y Epstein Barr negativo.
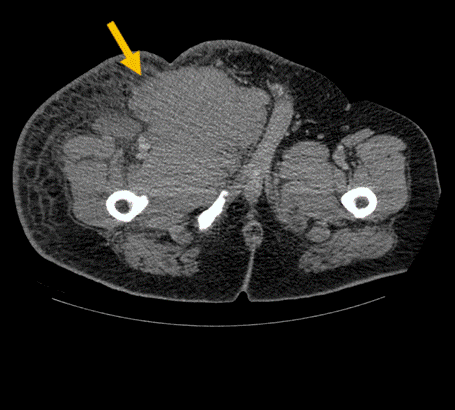
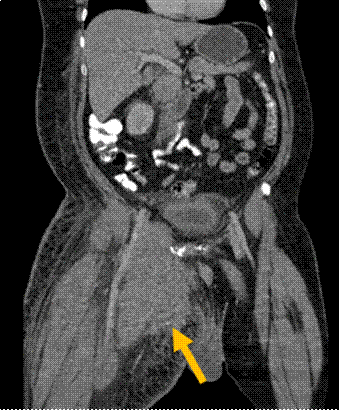
Se realiza tomografía computada de tejidos blando en la que se observa gran lesión con realce homogéneo del contraste que se extiende desde la región inguinal hasta la región anterior medial del muslo derecho proximal, asociado a cambios inflamatorios en tejidos blandos, de 20.2 x 10.5 x 15.2 cm en sus ejes longitudinal, antero posterior y transverso (Figuras 1 y 2).
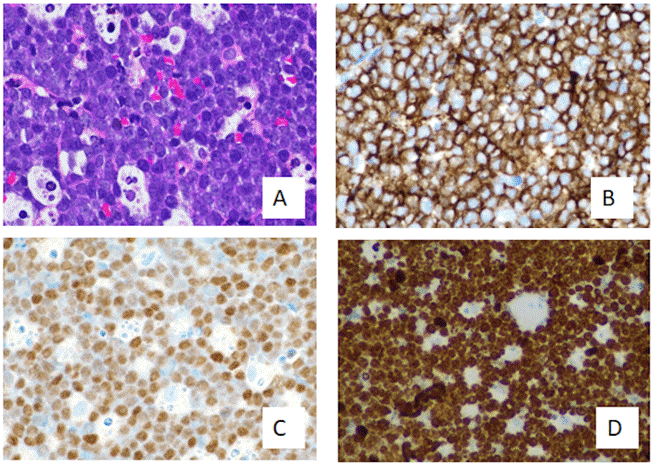
Se realiza biopsia por aguja gruesa por Radiología Intervencionista con resultado de patología (Figura 3) donde se observa patrón en cielo estrellado y linfocitos monótonos, de tamaño intermedio, con cromatina homogénea y nucléolos diminutos (A); positivas para expresión de CD20 y MYC (B y C respectivamente); con índice de proliferación elevado (ki-67) de 100% (D) y coexpresión de CD10 y BCL6 y negativos para BCL2, que sustenta el diagnóstico de linfoma de Burkitt.
Posteriormente presenta aumento de azoados, hiperuricemia, hiperfosfatemia e hipercalemia, sugestivo de síndrome de lisis tumoral espontáneo. Se inició manejo con hidratación; sin embargo, por mayor deterioro se ofrece terapia de reemplazo renal. Nuestra institución no cuenta con rasburicasa, el tratamiento de elección. Con evolución tórpida, requiriendo soporte ventilatorio y hemodinámico. Fallece a las 24 horas.
Discusión
El linfoma de Burkitt es un linfoma no Hodgkin de células B que se caracteriza por ser agresivo y de rápida replicación. Descrito por primera vez por el médico irlandés Denis P. Burkitt en una población pediátrica de Uganda(1).
Se divide en tres variantes: endémica, relacionada a inmunodeficiencia y esporádica. La variante endémica se caracteriza por su presencia en zonas endémicas de malaria, más relacionada a Plasmodium falciparum. Es común en la población pediátrica, afectando mandíbula, órbita o abdomen, siendo la afección del sistema nervioso central menor al 10%(1). La incidencia de la variable relacionada a la inmunodeficiencia es mayor en poblaciones entre 40 a 45 años. Se atribuye a 40% de los linfomas relacionados a la infección del virus de inmunodeficiencia humana (VIH)(1,2).
La variante esporádica tiene una presentación binodal, con alta incidencia a los 10 años y otra entre los 40 a 75 años. Los adultos varones son hasta 4 veces más afectados, sin predisposición racial o étnica. La principal localización de la variante esporádica es abdominal hasta un 80%, manifestándose con náuseas, vómitos, dolor, distensión abdominal y sangrado digestivo(2). Presenta afección al SNC del 20% y se asocia al virus de Epstein Barr hasta un 20%(1). A pesar de que se ha descrito la afección del sistema musculoesquelético por LNH, la afección de tejidos blandos por el linfoma de Burkitt es extremadamente rara, con pocos reportes de caso publicados(3,5). En este caso se observó una gran masa sólida a expensas de tejidos blandos que abarcaba el muslo derecho, sin compromiso intrabdominal.
Su presentación clínica es inespecífica; sin embargo, los síntomas más comunes son dolor asociado a la aparición de un tumor de rápido crecimiento(4). Éste se disemina rápidamente a ganglios linfáticos, sistema nervioso central y médula ósea. Al momento de su diagnóstico, la mayoría de los casos se encuentran en estadios avanzados (III y IV)(3).
La presentación patológica es similar en las tres variantes. La traslocación del MYC del cromosoma 8 al 14 t(8;14) es importante para la distinción del linfoma de Burkitt de otros linfomas de células B de alto grado y se encuentra presente en el 70-80% de los casos(1,5). Otras traslocaciones reportadas en menor porcentaje incluyen t(2;8) en aproximadamente 15% que involucra el gen de la cadena ligera kappa y t(8;22) en 5% relacionado a la cadena ligera lambda(6). La biopsia es distintiva por su presentación en cielo estrellado que representa el patrón de macrófagos que han ingerido restos celulares apoptósicos(1), como lo mostramos en la figura 3A. Otras neoplasias hematolinfoides que expresan este patrón son el linfoma difuso de células B grandes, linfoma de células B de alto grado, linfoma plasmablástico, linfoma de células grandes B ALK +, linfomas de células del manto (variantes agresivas) y leucemia/linfoma linfoblástico de células B o T(7).
Debido a la alta replicación tumoral, es común la presentación de síndrome de lisis tumoral espontáneo, que representa una emergencia oncometabólica que genera daños a órgano blanco debido a la muerte masiva de las células tumorales, como ocurrió en este caso. Sus manifestaciones son dependientes de la alteración hidroelectrolítica consecuente(1,2,5). Al ser identificada la urgencia se debe iniciar tratamiento temprano con hidratación intensa y alopurinol previo al inicio de quimioterapia; además se debe agregar rasburicasa como tratamiento profiláctico para la hiperuricemia(1); sin embargo, en nuestro medio no contamos con dicho medicamento.
El tratamiento inicia con dosis bajas de corticoides y posteriormente el régimen de quimioterapia conformado por etopósido, prednisona, vincristina, ciclofosfamida, doxorrubicina y rituximab (R-DA-EPOCH) y se inicia según estratificación de riesgo, tomando en cuenta estadío de la enfermedad, estado funcional, niveles de LDH y tamaño del tumor(1,2). La supervivencia puede llegar hasta 80% de obtener el diagnóstico temprano y así brindar tratamiento oportuno; así mismo, al retrasar tratamiento la mortalidad aumenta en estos casos(5).
Conclusión
La afección de tejidos blandos en miembros inferiores por linfoma de Burkitt es una manifestación extremadamente rara y atípica de la enfermedad. Su variante esporádica es infrecuente en adultos y queda en evidencia con este caso el pobre pronóstico que representa, incluso con el diagnostico oportuno. Su alto índice de proliferación lleva a complicaciones metabólicas, como el síndrome de lisis tumoral espontáneo, aumentando de manera considerable la mortalidad. Es de nuestro conocimiento que este caso sería el primero reportado en nuestra región.
Bibliografía
1. Roschewski M, Staud L, Wilson W. Burkitt’s Lymphoma. New England Journal of Medicine. 2022;387(12):1111–22.
2. Saleh K, Michot JM, Camara-Clayette V, Vassetsky Y, Ribrag V. Burkitt and Burkitt-Like Lymphomas: a Systematic Review. Curr Oncol Rep. 2020 Mar 6;22(4):33.
3. García-Barredo R, Fernández Echevarrí M, del Riego M, et al. Soft tissue Burkitt’s lymphoma: radiological findings. European Radiology. 1998;8(9):1654–1656.
4. Mechtoune M, et al. Pathological fracture of the knee revealing Burkitt lymphoma: a case report. PAMJ Clinical Medicine. 2021;5(81). https://10.11604/pamj-cm.2021.5.81.28830. [G1]
5. Mirrakhimov, Aibek E. Tumor Lysis Syndrome: A Clinical Review. World Journal of Critical Care Medicine. 2015; 4(2):130.
6. Kiefer T, Schüler F, Knopp A, et al. A human Burkitt's lymphoma cell line carrying t(8;22) and t(14;18) translocations. Ann Hematol. 2007;86(11):821-830.
7. Dy-Ledesma JL, Khoury JD, Agbay RL et al. Starry Sky Pattern in Hematopoietic Neoplasms: A Review of Pathophysiology and Differential Diagnosis. Adv Anat Pathol. 2016;23(6):343-355.
 |
 |
 |